
Reaction mechanisms and catalyst design
The main objective of the research within the “Catalysts for Hydrocarbons” group is to make contributions to the development of renewable carbon resources for societies energy needs in the transportation sector. Our investigations target the understanding of these processes on the atomic-scale through periodic density functional theory (DFT) and ab initio calculations on cluster models. Here we use higher-level methods (MP2 or DLPNO-CCSD(T)) since common DFT approaches typically underestimate barriers significantly1-3. The main program packages used are VASP, TURBOMOLE and ORCA. The main topics within the group are the conversion of methanol to hydrocarbons and the targeted functionalization of biomass as well as catalyst deactivation.
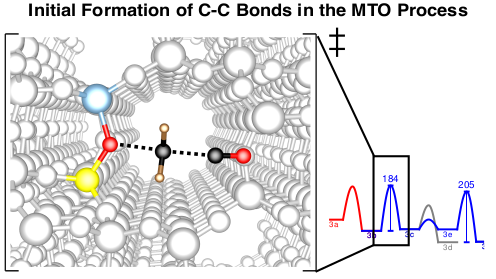
Methanol to hydrocarbons
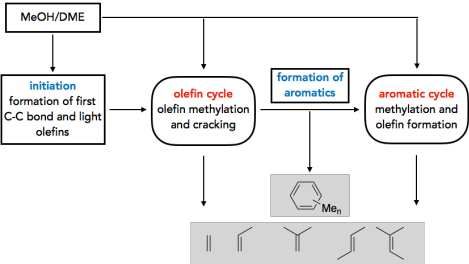
The methanol-to-olefins (MTO) and related processes allow the synthesis of hydrocarbons from biomass via the syngas-methanol route. There is thus an increased interest in a mechanistic understanding of the reactions that control selectivity and deactivation of the employed acidic zeotype catalysts. Another intriguing question concerns the initiation of the MTO process. While it is accepted that the reactivity is at later stages determined by catalytically active hydrocarbons that constitute the socalled hydrocarbon pool (HCP), it remains unclear, how the HCP is established. We tackle this problem by exploring a large number of potential initiation reactions, using accurate CCSD(T) and MP2 calculations on cluster models in addition to periodic DFT calculations1-3. Kinetic modelling shows that direct C-C bond formation from methanol is indeed a viable initiation mechanism and the most favorable reaction path proceeds via formation of CO from methanol followed by methylation.4
In addition to initiation, we investigate the role of olefins5 and aromatic molecules6 in the HCP.
Alternative approaches to fuels from renewable feed stocks
Another interesting class of products are oxymethylene dimethyl ethers (OMEs), which can be derived from methanol and formaldehyde. OMEs have shown great potential for the use as diesel fuel additives, but challenges for the catalytic process to produce targeted OMEs with the right chain-lengths remain. Most commonly, solid acid catalysts like zeolites are employed for these processes. Here we focus on the reaction of trioxane (TOX) and OME1 as alternative precursors. We find that direct incorporation of TOX is more favorable than its prior decomposition to formaldehyde, thus explaining the observed initial selectivity for OME47.
Metal-oxide interaction and catalyst deactivation
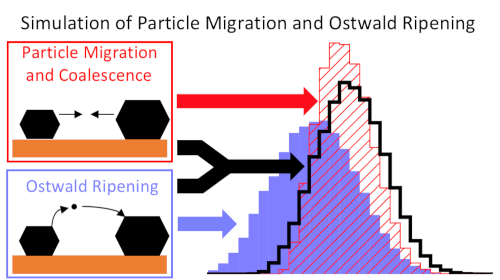
Another project of the group is catalyst deactivation, in particular through sintering. Here, we are currently focusing on the development of improved models for sintering8. Apart from classical, surface-mediated Ostwald ripening, we are particularly interested in ripening via the vapor phase. This mechanism is believed to be relevant for platinum, where the volatile species PtO2 may be formed under high temperatures and oxidizing conditions. Using mean-field models, we find that this mechanism is indeed realistic and can explain experimental sintering kinetics9. We furthermore study metal-oxide interaction through adhesion and the formation of ‘single atoms’.10-11
Publications
- Plessow, P. N.; Studt, F., Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations. ACS Catal. 2017, 7 (11), 7987-7994.
- Goncalves, T. J.; Plessow, P. N.; Studt, F., On the Accuracy of Density Functional Theory in Zeolite Catalysis. ChemCatChem 2019, 11 (17), 4368-4376.
- Plessow, P. N.; Studt, F., How Accurately Do Approximate Density Functionals Predict Trends in Acidic Zeolite Catalysis? J. Phys. Chem. Lett. 2020, 11 (11), 4305-4310.
- Plessow, P. N.; Smith, A.; Tischer, S.; Studt, F., Identification of the Reaction Sequence of the MTO Initiation Mechanism Using Ab Initio-Based Kinetics. J. Am. Chem. Soc. 2019, 141 (14), 5908-5915.
- Plessow, P. N.; Studt, F., Olefin methylation and cracking reactions in H-SSZ-13 investigated with ab initio and DFT calculations. Catal. Sci. Technol. 2018, 8 (17), 4420-4429.
- Fečík, M.; Plessow, P. N.; Studt, F., A Systematic Study of Methylation from Benzene to Hexamethylbenzene in H-SSZ-13 Using Density Functional Theory and Ab Initio Calculations. ACS Catal. 2020, 10 (15), 8916-8925.
- Goncalves, T. J.; Arnold, U.; Plessow, P. N.; Studt, F., Theoretical Investigation of the Acid Catalyzed Formation of Oxymethylene Dimethyl Ethers from Trioxane and Dimethoxymethane. ACS Catal. 2017, 7 (5), 3615-3621.
- Dietze, E. M.; Abild-Pedersen, F.; Plessow, P. N., Comparison of Sintering by Particle Migration and Ripening through First-Principles-Based Simulations. J. Phys. Chem. C 2018, 122, 26563−26569.
- Plessow, P. N.; Abild-Pedersen, F., Sintering of Pt Nanoparticles via Volatile PtO2: Simulation and Comparison with Experiments. ACS Catal. 2016, 6 (10), 7098-7108.
- Goodman, E. D.; Johnston-Peck, A. C.; Dietze, E. M.; Wrasman, C. J.; Hoffman, A. S.; Abild-Pedersen, F.; Bare, S. R.; Plessow, P. N.; Cargnello, M., Catalyst deactivation via decomposition into single atoms and the role of metal loading. Nat. Catal. 2019, 2 (9), 748-755.
- Dietze, E. M.; Plessow, P. N., Predicting the Strength of Metal–Support Interaction with Computational Descriptors for Adhesion Energies. J. Phys. Chem. C 2019, 123 (33), 20443-20450.